| Version 58 (modified by , 16 years ago) (diff) |
|---|
SNP calling pipeline
Status: Alpha Authors: Freerk van Dijk, Morris Swertz
This is the documentation of the BBMRI-NL snp calling pipeline based on the Broad GATK. It consists of the following three workflows:
- Workflow 1: SnpCallingPipeline/ReferencePreparation
- Workflow 2: SnpCallingPipeline/AlignmentAndCleaning
- Workflow 3: SnpCallingPipeline/VariantCalling
Schematic Overview
This simplified overview this schema hides intermediate sort and indexing steps and only shows data inputs/outputs first time they occur.
Discussion
- How long takes alignment per genome?
- If this takes very long we can split read files
- How long takes realign knownsonly (per genome)?
- If very long, we need to rewrite workflow 2 to split before realign
- For realign: if we split per chromosome, can we also split bam file?
- How to easily lift over from b36 to b37
- Contact BGI if they can use b37??
Todo:
First:
- Recode workflow 2 to work per genome instead of per chromosome and test - Freerk (done) -> workflow3 still needs to be done
- Run on pilot data (6) to evaluate timing and concurrency issues (can 6 run on one node?) - Freerk (in progress)
- Complete analysis of data (60) until including merge to sample.aligned.bam - Freerk
- QC pipeline - Can we get Jeroen and Yurii involved here
- UnifiedGenotyper? without realign - Freerk
- GATK variant eval to make venn diagrams
- Contact Yurii for this; Let Jeroen take charge? (done) -> Jeroen doing QC stuff
- Share data with Grid following plan Silvia - Freerk
- Contact BGI for sample list - Morris (done)
- Put report on FTP - Ger,Freerk
Next:
- Short tutorial howto generate pipeline scripts - Morris
- Teach Barbara and Jeroen
- Port pipeline to Grid with help of Barbara
- What do we need to generate exactly - Barbara
Optimization?
Current
Step Cores Memory (gb) Time (hh.mm)
BWA alignment 1 ± 6 10.05
BWA spe 1 3.35
Sam-Bam 1 12.3
Sam sort 1 5.05
Mark Duplicates 1 4 1.55
Realignment (knowns only) 1 8 (*can be lowered) 5.2
Fix mates 1 6 (*) 3.05
Covariates bef. 1 2 12.35
Recalibrate 1 4 7.3
Sam sort 1 4.5
Covariates aft. 1 2 11.2
Analyze Covar. 1 4 < 00.01
Disk
- Option 1, If it is possible to let a node guarantee certain amount of disk space (/tmp), we should use the entire cluster. Before start running a pipeline, we can just ask the node to reserve that amount of disk space.
- Option 2, If we can cut a dedicate part of the cluster, we can use our own scheduler to share the nodes/disks. E.g, depending on the disk space usage pattern and how we can remove the data, we can decide which jobs run at which node and when.
Memory/CPU time
- Can multiple samples use the same reference genome in memory during the BWA alignment. I.e. 1 sample->6GB, 3 samples->6GB.
- NO?
- Can we parallelize the Markduplicate?
- YES!
- MarkDuplicates finds sequence pairs that map to the same position, marking or removing the duplicates so you can work with unique pairs in downstream analyses. If you want them removed, use the REMOVE_DUPLICATES=true flag when running the program.
- Can we parallelize covariate before/after, recalibration?
- Don't know
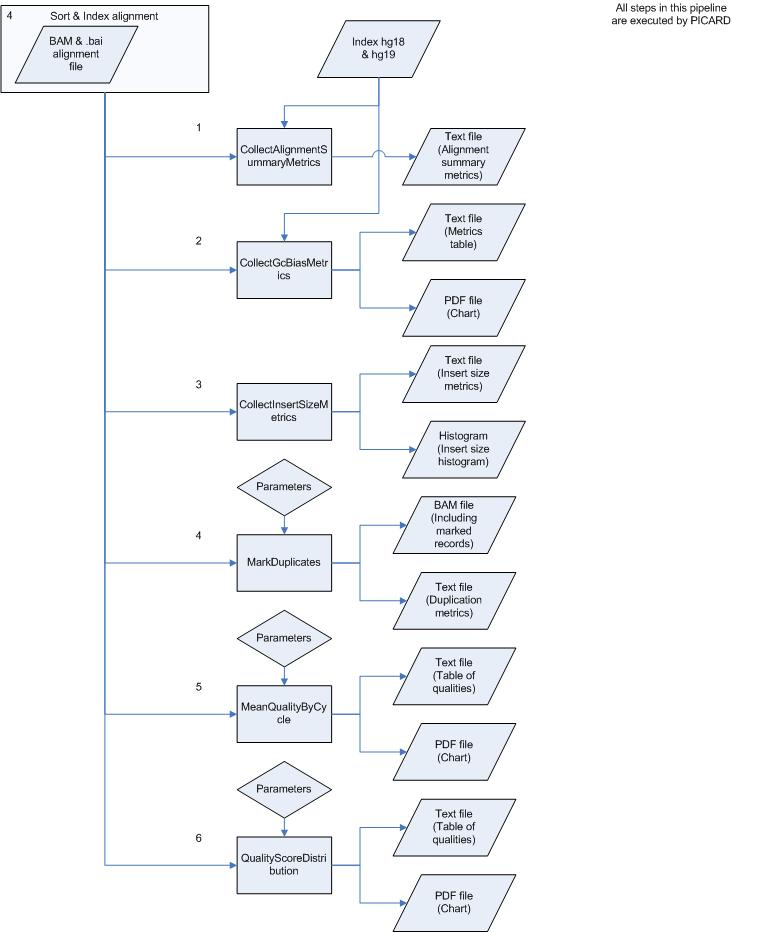
List of steps
Attachments (3)
- Figure1.png (349.2 KB) - added by 16 years ago.
- Figure2.png (311.5 KB) - added by 16 years ago.
- Figure3.png (224.0 KB) - added by 16 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip